A multiplatform metabolomics/reactomics approach as a powerful strategy to identify reaction compounds generated during hemicellulose hydrothermal extraction from agro-food biomasses

- Photo: Food Chemistry Volume 421, 2023, 136150: Fig. 1. Representation of samples preparation before 1H NMR, GC–MS, and UHPLC-IM-Q-TOF-MS analysis.
In the research article published in the Food Chemistry journal, the researchers from the University of Parma, Italy, the Wageningen University & Research, the Netherlands, and Ferrero Group, Italy explored the metabolomics of degradation compounds formed during hydrothermal treatment of hazelnut shells, a process commonly used for hemicellulose extraction.
Using a combination of GC–MS, ¹H NMR, and UHPLC-IM-Q-TOF-MS, the researchers identified and quantified various organic acids, modified sugars, and aromatic compounds, alongside furans, polyols, N-heterocyclic compounds, aldehydes, ketones, and esters. The innovative use of ion mobility-based LC-MS enabled the detection of molecules with diverse molecular weights and offers the potential for building databases linking compound formation to processing conditions. This approach holds promise for risk assessment and optimizing hydrothermal processes for lignocellulosic materials.
The original article
A multiplatform metabolomics/reactomics approach as a powerful strategy to identify reaction compounds generated during hemicellulose hydrothermal extraction from agro-food biomasses
Andrea Fuso, Laura Righetti, Franco Rosso, Ginevra Rosso, Ileana Manera, Augusta Caligiani
Food Chemistry Volume 421, 2023, 136150
https://doi.org/10.1016/j.foodchem.2023.136150.
licensed under CC-BY 4.0
Selected sections from the article follow. Formats and hyperlinks were adapted from the original.
Highlights
- Hydrothermal treatment of hazelnut shells was carried out.
- The pattern of degrading compounds was investigated combining three techniques.
- Organic acids, modified sugars and aromatic compounds were detected and quantified.
- UHPLC-IM-QTOF-MS allowed to putatively identify 212 compounds, even with high Mw.
- HRMS is a candidate for untargeted metabolomics approaches on these extracts.
Abstract
Hydrothermal treatment is commonly used for hemicelluloses extraction from lignocellulosic materials. In this study, we thoroughly investigated with a novel approach the metabolomics of degradation compounds formed when hazelnut shells are subjected to this type of treatment. Three different complementary techniques were combined, namely GC–MS, 1H NMR, and UHPLC-IM-Q-TOF-MS. Organic acids, modified sugars and aromatic compounds, likely to be the most abundant chemical classes, were detected and quantified by NMR, whereas GC- and LC-MS-based techniques allowed to detect many molecules with low and higher Mw, respectively. Furans, polyols, N-heterocyclic compounds, aldehydes, ketones, and esters appeared, among others. Ion mobility-based LC-MS method was innovatively used for this purpose and could allow soon to create potentially useful datasets for building specific databases relating to the formation of these compounds in different process conditions and employing different matrices. This could be a very intelligent approach especially in a risk assessment perspective.
1. Introduction
In the last decades much attention has been paid in combatting the phenomenon of food waste, which represents a big issue for ethical, environmental, and economic reasons. Among all foodstuffs, it is well known that products of vegetable origin, such as cereals, tubers, roots, fruit, and vegetables, are connected to great quantities of waste. The composition of these wastes can be disparate, but according to a recent study about 90% of plant biomass is represented by lignocellulosic material (Saini, Saini, & Tewari, 2015), which means that it is mainly composed of lignin, hemicellulose and cellulose, in variable proportions. The valorization of these by-products, due to the extraordinary structural complexity of the polymers present within them, cannot disregard the principle of biorefinery, i.e. the separation of the individual biomass fractions for the production of different components, such as biomolecules, biomaterials, bioenergy and biofuels (Villacís-Chiriboga, Vera, Van Camp, Ruales, & Elst, 2021). In a historical period in which consumer’s food choices are changing, moving towards healthier foods, dietary fibres naturally play a fundamental role. For this reason, hemicelluloses in particular acquire an even greater global interest. Indeed, hemicelluloses have a wide variety of applications: for instance, they can be easily transformed into oligosaccharides with interesting bio-functional properties or further depolymerized into pentoses and hexoses for subsequent conversion into bioethanol and chemical substances (Wang et al., 2016). Hemicelluloses are relatively small heteropolysaccharides (degree of polymerization 100–200 units), branched and made of five- and six-carbon monosaccharide units, of which the most frequent are xylose, mannose, arabinose, galactose, glucose, and glucuronic acid, as well as acetyl groups (Luo et al., 2019). These complex polymers were found to be linked by hydrogen bonds and van der Waals interactions with cellulose, forming highly resistant networks (Luo et al., 2019), and at the same time they interact with lignin through complex interactions, which are radical coupling of ferulate substitutions and incorporation of hemicellulosic glycosyl residues by re-aromatization of lignification intermediates (Terrett & Dupree, 2019). Hence, to extract and isolate hemicelluloses, “strong” methods are needed, and the most used combine high temperatures and high pressures. The most common in this sense is the hydrothermal treatment (HT), also called autohydrolysis treatment, which consists in subjecting the matrix of interest to extraction at 160–220 °C in water, kept in the liquid state thanks to the high pressures (Kumari & Singh, 2018). When such treatment is applied, however, it is very likely to get the formation in the reaction medium of innumerable undesired compounds, such as degradation products derived from lignin, sugars, or proteins, as well as monosaccharides, furans and organic acids. The mechanism of formation of these compounds is extremely complicated, since it is strictly related to the time/temperature conditions set, to the pH and to the matrix. Depending on the reaction conditions, for example, glucose can be converted into 5-(hydroxymethyl)-2-furaldehyde (HMF) and/or levulinic acid, formic acid and various phenolics at high temperatures, while xylose can follow different reaction mechanisms originating furan-2-carbaldehyde (furfural) and/or various C-1 and C-4 compounds (Rasmussen, Sørensen, & Meyer, 2014). Furthermore, monosaccharides can further react to form pseudo-lignin, humine, aldehydes, ketones, organic acids, or aromatic compounds (Rasmussen, Sørensen, & Meyer, 2014). Several studies have shown how it was possible to vary the composition of the autohydrolysis liquor after HT in different conditions, mainly in terms of furfural, HMF, acetic, formic, and lactic acid content (Wang et al., 2016). At the same time, other research has pointed out that although some mechanisms are to date well understood, many other metabolic pathways remain unknown (Gao et al., 2016). Therefore, since one of the main aims of reusing hemicellulose from vegetable by-products is its transformation into healthy ingredients for food companies, it is of enormous importance to further investigate the presence and the formation of all these compounds that originate following HT, especially to evaluate them in terms of potential toxicity. This investigation must be done both through the study and understanding of the reaction mechanisms, and through the improvement of analytical techniques that allow their identification and quantification. In this complex scenario, where multiple reaction pathways not fully understood led to very complicated mixtures of neoformed compounds, there is the need of combining different and complementary analytical approaches, to further unravelling the reaction mechanisms and better characterize the composition of lignocellulose hydrothermal extracts. To this aim, in the present work the molecular composition of the hydrothermal extracts of hazelnut shells (HS) was studied using different metabolomics platform, namely 1H NMR, GC–MS and UHPLC-IM-Q-TOF-MS.
2. Materials and methods
2.1. Chemicals and reagents
HPLC-grade acetonitrile, ammonium formiate, trifluoroacetic acid (TFA), N,O-Bis(trimethylsilyl)trifluoroacetamide (BSTFA), 3-(trimethylsilyl)propionate-d4 (TSP), d-glucose, d-fructose, d-galactose, d-mannose, d-rhamnose, d-ribose, d-xylose, d-fucose, d-galacturonic acid, d-glucuronic acid, d-glucosamine, d-galactosamine and phenyl-β-d-glucopyranoside were purchased from Sigma-Aldrich (Taufkirchen, Germany); bidistilled water was obtained using Milli-Q System (Millipore, Bedford, MA, USA); diethyl ether, hydrochloric acid and ethanol were purchased from Carlo Erba (Milan, Italy); D2O was bought from VWR (Radnor, PA, USA); MS-grade formic acid from Fisher Chemical (Thermo Fisher Scientific Inc., San Jose, CA, USA) was also used. Leucine-enkephalin [186006013] used as lock mass solution and Major Mix IMS/TOF Calibration Kit [186008113] for mass and CCS calibration were purchased from Waters (Manchester, UK).
2.2. Hydrothermal treatment of hazelnut shells
Hazelnut shells (HS) were kindly provided by Ferrero S.p.A. (Cuneo, Italy), finely ground and sieved with grain size <500 µm. The hydrothermal treatment (HT) was performed in a stainless-steel Parr reactor 4566 (Parr Instrument Company, Moline, Illinois, USA) with an internal volume of 300 mL and internal cooling loop, equipped with Parr PDI for temperature control (model 4848). 5 g of ground sample were extracted with 125 mL of bidistilled water, and HT was carried out under isothermal conditions for 60 min at 175 °C. The time to reach the working temperature was 30 min and the final relative pressure was 8.1 bar. After treatment, the obtained solid and liquid phases were separated by centrifugation at 3900 rpm for 30 min at 4 °C. Finally, the supernatant was collected and freeze-dried. All the following analyses described in the next sections were performed on this sample.
2.3. Proximate composition of HS extract
Proximate composition of HS extract was investigated using standard procedures (AOAC, 2002). Moisture was determined by drying in oven at 105 °C for 24 h. Total ash was determined through mineralization at 550 °C in two steps, each one lasting 5 h. Total nitrogen was determined with a Kjeldahl system (DKL heating digester and UDK 139 semiautomatic distillation unit, VELP SCIENTIFICA) using 6.25 as a nitrogen-to-protein conversion factor. Lipid content was determined using a Soxhlet extractor (SER 148/3 VELP SCIENTIFICA, Usmate Velate, Italy) employing diethyl ether as extracting solvent.
For the determination of total sugars and monosaccharide distribution, 10 mg of sample were dissolved in 3 mL of 2 N trifluoroacetic acid (TFA) and hydrolysed at 110 °C for 2 h under stirring. Then, 900 µL of the solution were withdrawn and put together with 150 µL of 1000 ppm phenyl-β-d-glucopyranoside, used as internal standard, and then evaporated by rotavapor. The obtained dried hydrolysate was washed with 1 mL of methanol to remove the residue of TFA and evaporated again. 1 mL of 0.5 M NH4OH was subsequently added to delactonize the eventually present acid sugar lactones in the sample, and again evaporated by rotavapor. Finally, the dried hydrolysate was dissolved in 800 µL of dimethylformamide (DMF) and 200 µL of N,O-Bis(trimethylsilyl)trifluoroacetamide (BSTFA), the latter used as derivatizing agent. The reaction was held for 1 h at 60 °C and finally the derivatized sample was injected in gaschromatography. GC–MS analysis of monosaccharides was performed with a 6890 N gas chromatograph coupled to a 5973 N mass selective detector (Agilent technologies, Santa Clara, CA). A SLB-5 ms, 30 m × 0.25 mm, 0.25 µm thickness column (Supelco, Bellafonte, PA, USA) was used. The chromatogram was recorded in the scan mode (40–500 m/z) with a programmed temperature from 60 °C to 270 °C. The initial temperature was 60 °C, held for 2 min, then increased to 160 °C at a rate of 10 °C/min, held isothermal for 5 min, increased to 220 °C at a rate of 10 °C/min, kept for 5 min, increased to 270 °C at a rate of 20 °C/min and maintained for 5 min. Quantification was performed with a response factor, considering the area and concentration ratios between the internal standard (phenyl-β-d-glucopyranoside), and the following monosaccharides: d-glucose, d-fructose, d-galactose, d-mannose, d-rhamnose, d-ribose, d-xylose, d-fucose, d-galacturonic acid, d-glucuronic acid, d-glucosamine and d-galactosamine.
2.4. Gaschromatography-mass spectrometry (GC–MS) analysis
The protocol for samples preparation before GC–MS analysis is represented in Fig. 1. Briefly, 300 mg of sample were weighted and added to 6 mL of water and the extraction was carried out at 50 °C for 2 h. Later, the sample was split in three different 2 mL aliquots: the first (“H2O”) was simply analyzed after putting 200 µL in a round-bottomed flask containing 65 µL of 1160 ppm phenyl-β-glucopyranoside, used as standard. In both the second and third aliquot, 8 mL of ethanol were firstly added to make the fibres to precipitate. The solutions were thoroughly mixed and centrifuged at 4 °C and 3900 rpm for 25 min. Later, in the second aliquot (“EtOH”) about 1700 µL of the supernatant were withdrawn and put together with 65 µL of 1160 ppm phenyl-β-glucopyranoside in a new round-bottomed flask. In the third aliquot (“HCl”), 3 mL of the supernatant obtained after centrifugation were withdrawn, put in a new round-bottomed flask, and dried by rotavapor, then here 2 mL of 4 N HCl were added, and the solution was allowed to stand at 100 °C for 3 h under stirring. At the end of the reaction, the same amount of phenyl-β-glucopyranoside used in the other experiments was added. Finally, the three solutions were dried by rotavapor, then derivatized by adding 800 µL of DMF and 200 µL of N,O-Bis(trimethylsilyl)trifluoroacetamide (BSTFA), the latter used as derivatizing agent, and allowing the reaction to stand for 1 h at 60 °C before being injected in GC–MS.
 Food Chemistry Volume 421, 2023, 136150: Fig. 1. Representation of samples preparation before 1H NMR, GC–MS, and UHPLC-IM-Q-TOF-MS analysis.
Food Chemistry Volume 421, 2023, 136150: Fig. 1. Representation of samples preparation before 1H NMR, GC–MS, and UHPLC-IM-Q-TOF-MS analysis.
GC–MS analysis of the degradation products originated from the HT was performed with a 6890 N gas chromatograph coupled to a 5973 N mass selective detector (Agilent technologies, Santa Clara, CA). A SLB-5 ms, 30 m × 0.25 mm, 0.25 µm thickness column (Supelco, Bellafonte, PA, USA) was used. The mass spectrometer operated in the electron impact (EI) ionization mode (70 eV) and the ion source temperature was set at 230 °C. The chromatogram was recorded in the scan mode (50–800 m/z) with a programmed temperature from 60 °C to 270 °C. The initial temperature was 60 °C, held for 2 min, then increased to 160 °C at a rate of 10 °C/min, held isothermal for 10 min, increased to 220 °C at a rate of 10 °C/min, kept for 5 min, increased to 270 °C at a rate of 20 °C/min and maintained for 15 min. A semi-quantification of each analyte identified was performed, according to the equation (1) below:
(1) \(\text{ppm}_{\text{analyte}} = \frac{\text{Area}\text{analyte*ppm}\text{phenyl -}\beta \text{- D - glucopyranoside}}{\mathrm{Area}phenyl - \beta - D - \text{glucopyranoside}}\)
The identification of the various compounds was performed comparing mass spectra with WILEY library data.
2.5. 1H NMR analysis
An aqueous extract of the freeze-dried extract obtained after hydrothermal treatment of HS was analysed through 1H NMR. Briefly, 200 mg of sample were added to 2 mL D2O together with 100 µL of 2210 ppm TSP dissolved in D2O, and the extraction was performed under stirring at 50 °C for 2 h. Then, the solution was centrifuged at 4 °C and 3900 rpm for 25 min and subsequently filtered through a 0.45 µm nylon membrane in a 5 mm NMR tube (Fig. 1).
1H NMR spectra were recorded on a Bruker Avance III 400 MHz NMR Spectrometer (Bruker BioSpin, Rheinstetten, Karlsruhe, Germany) operating at a magnetic field-strength of 9.4 T. A 1D 1H NOESY sequence, previously optimized to remove the residual water signal with minimal affection of the baseline, was utilized for water suppression (Musio et al., 2020). Spectra were acquired at 298 K, with 32 K complex points, using a 90° pulse length and 5 s of relaxation delay (d1). 128 scans were acquired with a spectral width of 20 ppm, an acquisition time of 1.707 s, 8 dummy scans and a mixing time of 0.010 s. The complete relaxation of the protons obtained during acquisition time and relaxation delay allowed to use integrals for quantitative purposes. Different spectral zones were identified and assigned to diverse classes of compounds found in hydrothermal extracts of hazelnut shells. The same zones were integrated and used for a preliminary crude quantification of the different compounds. Acetyl groups deriving from free acetate and substituted sugars (mainly acetylated xylans) were quantified integrating the spectral zone 1.92–2.34 ppm. Total aromatic compounds were determined from the integration of the whole zone 5.97–7.78 and expressed as phenylpropanoids. The signal of formic acid at 8.45 was integrated as determinant indicative of a part of degraded sugars according to the mechanism of levulinic acid formation (Pyo, Glaser, Rehnberg, & Hatti-Kaul, 2020).
2.6. Ultra-high-performance liquid chromatography – ion mobility – Quadrupole time-of-flight – mass spectrometry (UHPLC-IM-Q-TOF-MS) analysis
The reaction products originated from hydrothermal treatment of HS were further identified through UHPLC-IM-Q-TOF-MS. The sample was simply dissolved in water and analysed (Fig. 1). Specifically, 1 mg of the freeze-dried extract obtained after HT was dissolved in 1 mL of MilliQ water.
2.6.1. UHPLC-TWIMS-QTOF analysis
ACQUITY I-Class UHPLC separation system coupled to a VION IMS QTOF mass spectrometer (Waters, Wilmslow, UK) equipped with an electrospray ionization (ESI) interface was employed. Samples were injected (2 µL) and chromatographically separated using a reversed-phase ACQUITY Premier HSS T3 column 2.1 × 100 mm, 1.8 µm particle size (Waters, Milford, MA, USA). A gradient profile was applied using water 5 mM ammonium formiate (eluent A) and acetonitrile (eluent B) both acidified with 0.1% formic acid as mobile phases. Initial conditions were set at 2% B, after 3 min of isocratic step, a linear change to 100% B was achieved in 12 min and holding for 5 min to allow for column washing before returning to initial conditions. Column recondition was achieved over 3 min, providing a total run time of 23 min. The column was maintained at 45 °C and a flow rate of 0.35 mL/min used.
Mass spectrometry data were collected in positive and negative electrospray mode over the mass range of m/z 50–1100. Source settings were maintained using a capillary voltage, 2.5 kV and 2 kV for positive and negative ESI modes, respectively; source temperature, 150 °C; desolvation temperature, 500 °C and desolvation gas flow, 950 L/h. Lock mass correction was performed by infusing a solution of leucine-enkephalin [M + H]+ (m/z 556.2766, calibration kit from Waters) at a concentration of 200 pg/µL (infusion rate 10 µL/min) and acquired every 2.5 min to provide a real-time single-point mass calibration. The TOF analyzer was operated in sensitivity mode and data acquired using HDMSE, which is a data independent approach (DIA) coupled with ion mobility. Low-energy scan (CE 6 V) and a high-energy scan (CE ramp 15–50 V) were alternatively acquired during the run. The optimized ion mobility settings included: nitrogen flow rate, 90 mL/min (3.2 mbar); wave velocity 650 m/s and wave height, 40 V. Device within the Vion was calibrated using the Major Mix IMS calibration kit (Waters, Wilmslow, UK) to allow for collisional cross section (CCS) values to be determined in nitrogen. The calibration covered the CCS range from 130 to 306 Å2. The TOF was also calibrated prior to data acquisition and covered the mass range from 151 Da to 1013 Da. TOF and CCS calibrations were performed for both positive and negative ion mode. Data acquisition was conducted using UNIFI 1.8 (Waters, Wilmslow, UK) which enable to generate CCS values for all the detected species from the automatic CCS calibration.
2.6.2. Data processing
Data processing and compound identification were conducted using Progenesis QI Informatics (Nonlinear Dynamics, Newcastle, UK). Each UHPLC-MS run was imported as an ion-intensity map, including m/z and retention time, that were then aligned in the retention-time direction (0–20 min). From the aligned runs, an aggregate run representing the compounds in all samples was used for peak picking. This aggregate was then compared with all runs, so that the same ions were detected in every run. Isotope and adduct deconvolution were applied, to reduce the number of features detected. Metabolites were identified by publicly available database searches including PubChem as well as by fragmentation patterns, using Progenesis QI.
3. Results and discussion
3.1. Proximate composition of HS extract
After undergoing the HT and before studying the degradation compounds originated from it, both proximate composition and monosaccharide distribution of the freeze-dried extract was investigated. Results are reported in Table 1.
 Food Chemistry Volume 421, 2023, 136150: Table 1. Proximate composition and fibre’s monosaccharide distribution of the freeze-dried extract obtained following ht of hs.
Food Chemistry Volume 421, 2023, 136150: Table 1. Proximate composition and fibre’s monosaccharide distribution of the freeze-dried extract obtained following ht of hs.
As expected, this sample was predominantly made of sugars, which accounted for half of its fresh weight (see Table 1). As concerns monosaccharide distribution of the fibres extracted, they were mainly made of xylose (63%). Other sugars, such as arabinose and glucuronic acid, were also present probably as substituents along the xylose-based chain, as often happens in xylans extracted from lignocellulosic matrices. Finally, galacturonic acid and glucose could be derived from pectin and cellulose fractions, while rhamnose and galactose could derive from pectin as well.
A relevant amount of ash, equal to 9%, was also found, while proteins and lipids were quite lower (3% and 1%, respectively). Interestingly, the sum of moisture, proteins, lipids, ash, and total sugars accounted for only 65%. By 1H NMR analysis it was also possible to investigate the amount of acetyl groups attached to the xylan backbone, and to estimate both the aromatic compounds, derived from lignin and calculated as phenylpropanoids, as well as the sugars chemically modified by thermal degradation, overall reaching a total sum of about 80%. However, about one-fifth of the percentage by weight of the gross composition remained uncharacterized and most likely constituted by neoformation compounds risen from polysaccharides and lignin during the thermal process. In the effort of characterizing as much as possible these process-derived compounds, different analytical methods have been applied, as reported in the following paragraphs.
3.2. GC–MS analysis
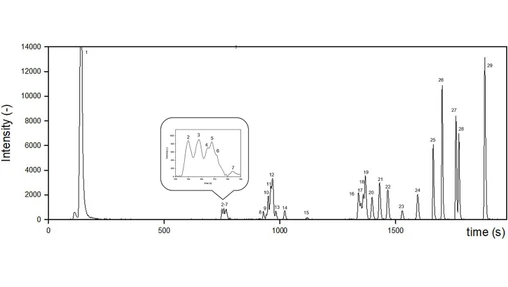
Following GC–MS analysis carried out on the sample treated in three different ways (Fig. 1), compounds identification was carried out by comparing the mass spectra of the trimethylsilyl (TMS) derivatives with Wiley275 library. A total of 54 compounds were selected and integrated as they are likely to be derived from HT (Table 2).
 Food Chemistry Volume 421, 2023, 136150: Table 2 (truncated). List of compounds originated after ht, identified and semi-quantified by gc–MS.
Food Chemistry Volume 421, 2023, 136150: Table 2 (truncated). List of compounds originated after ht, identified and semi-quantified by gc–MS.
As reported in Table 2, the set of identified and semi-quantified compounds constituted 1.4%, 2.3% and 5.9% of the initial sample when semi-quantified in the simple aqueous extract (H2O), in the aqueous extract followed by precipitation of the fibre by ethanol (EtOH), and in the aqueous extract followed by fibre precipitation and acid hydrolysis (HCl), respectively. It is important to emphasize how the acid hydrolysis carried out on the supernatant after fibre precipitation allows to quantify a total content of metabolites much higher than that obtained with a simple aqueous extract. This indicates the complex nature of the compounds extracted from the raw material or originated after the hydrothermal treatment, which are probably present conjugated to other compounds and not directly detectable by GC–MS due to the high molecular weight (Mw). The compounds were putatively identified comparing their mass spectra with Wiley 275 library and are reported in Table 2. They can be categorized into 5 main classes, namely organic acids, aromatic compounds, modified sugars (these three are the classes identified with 1H NMR technique as well), fatty acids and polyols. Then, other compounds were also detected, including a couple of N-heterocyclic compounds and various unidentified signals. Organic acids constituted, in terms of percentage of the initial sample, a rather small quantity, varying between 0.05% and 0.3%. In all three extracts, the most abundant ones were found to be lactic acid and glycolic acid, originating from glucose or xylose as a result of high temperatures (Onda, Ochi, Kajiyoshi, & Yanagisawa, 2008). Levulinic acid, that has been classified as one of the top 12 promising bio-based building blocks for the synthesis of fuels and chemicals, was found in very small concentrations, and this is not surprising since it is formed from hexoses or directly from cellulose (Pyo et al., 2020), which are quite scarce in our extract. On the other hand, the higher amount of formic acid quantified by 1H NMR could indicate the formation of a larger amount of levulinic acid which is further degraded during thermal treatment. 3-hydroxypropionic (3-HP) acid was found in very low concentrations too, and although its main origin is from glucose fermentation, in this case the formation is likely attributable to the oxidation of aromatic compounds (Shende & Levee, 1999). Finally, 5-hydroxyvaleric acid has been reported to derive from oxidation of furfural derivatives, such as cyclopentanone or 1,5-pentanediol, and therefore from xylose (Asano, Takagi, Nakagawa, Tamura, & Tomishige, 2019), while 2,3-dihydroxypropanoic acid (glyceric acid) is derived from glycerol oxidation (Yan et al., 2021) and 2-hydroxyhexanoic acid from sugar degradation in acid condition (Esteves et al., 2022). Even not detected by GC–MS, acetic and formic acids were also present, as evidenced by 1H NMR, originating from autohydrolysis of acetylated xylans and degradation of hexose, respectively. As a whole, the majority of organic acids detected, originated from sugar modification pathways.
Aromatic compounds as a class constituted a significant fraction of the extract, if compared to organic acids: their total quantity was indeed semi-quantified with values ranging from 0.4% to 1.1%. These compounds are formed from the aromatic residues of lignin and are degraded to many types of phenolic structures, depending on the monomeric units in the native lignin (Almeida et al., 2007). In fact, lignin is a polymer made of various amounts of three different monolignols, that are p-coumaryl alcohol, coniferyl alcohol, and sinapyl alcohol. For this reason, most of the identified aromatic compounds might be divided in three subclasses, namely p-coumaryl, coniferyl, or sinapyl alcohol derivatives.
Previous studies showed how the increase in temperature during HT leads to methoxy group (-O-CH3) hydrolysis with the consequent increase in p-coumaryl derivatives, making it harder to identify the mechanism of formation (Ramachandran et al., 2020). Among the compounds identified in our work by GC–MS, four of them were classifiable as coniferyl alcohol derivatives: 3-vanilyl propanol, vanilethanediol, hydroferulic acid and vanillylmandelic acid. The latter two were always found to be among the compounds present in the highest amount, even though not detected in “HCl” sample. The most abundant aromatic compound turned out to be benzoic acid, one of the most known lignin-derived molecules, and this may be explained by its great stability to high temperatures in liquid water (Dunn, Burns, Hunter, & Savage, 2003), while gallic acid, 7,8-dihydroxyflavone and diethylstilbestrol are likely derived from the raw material and were found only or especially in “HCl” sample, suggesting their presence in a complex with other structures.
Polyols were always found, as sum, in rather similar quantities in the three samples and varied between 0.3% and 0.5% of the initial sample, indicating their presence mainly as free form in the extract. An exception was glycerol, the most abundant one, its presence could be due to the thermal hydrolysis of triacylglycerols in HS and it is not surprising that the highest amount has been found in “HCl” sample, being even easier to release it following acid hydrolysis. Sorbitol probably derived from the reduction of glucose, while the pentitolsxylitol, arabitol and ribitol from the reduction of xylose and arabinose (Heuel et al., 1998, Verduyn et al., 1985) in the pentose and glucuronate interconversions pathway in the raw material. Erythritol has been reported as a compound mainly produced from glucose through yeast fermentation (Kobayashi & Fukuoka, 2013) while inositol, on the other hand, may be produced from the hydrolysis of phytic acid, and it has been recently demonstrated how its degradation through HT is effective already with low temperatures (Sheikh & Saini, 2022). Then, many fatty acids were found, only in HCl sample because deriving from hydrolysis of triglycerides present in the raw material, and among them the most abundant was palmitic acid. Their total amount correspond to about 0.5% of the initial sample and adding up this value to glycerol’s one the result turns out to be in accordance with the total lipid content reported in Table 1. One of the most numerous classes was that of modified sugars, that was the greatest in terms of abundance as well, reaching 4% of the initial sample in “HCl”, showing again the complex nature of these compounds, likely bound to each other and detectable only after acid hydrolysis. Some rare sugars, namely 2-O-methyl xylose and 2-O-methyl glucose were also found: the first may have been obtained as a result of a simultaneous extraction of pectin fraction present in HS, because it has been reported as a rhamnogalacturonan-II component (Reuhs et al., 2004). Glucuronic acid lactone can be obtained from degradation of d-glucuronic acid when subcritical water is used, while a lot of sugar-derivative peaks appeared especially in “HCl” sample, in the last part of the chromatographic run, but without being able to identify them. These can be released from the matrix after hydrolysis, but it is not excluded their ex-novo formation during acid treatment. Finally, regarding “other” compounds, not classifiable in any of the previous classes, different abundant compounds were found. The maximum quantity of this substances peaked to 0.2% of the freeze-dried extract obtained from HT of HS. Both 3-hydroxy-pyridine and 2,4(1H,3H)-Pyrimidinedione, dihydro-1,3-dimethyl can be originated from Maillard reaction (Yu-Chiang, Chi-Tang, & Hartman, 1992).
Despite gas chromatographic methods are very often used for the characterization of HT degradation compounds (Rasmussen et al., 2014), their indisputable limits related to the necessity of matrix hydrolysis and analyte derivatization hinder a complete molecular characterization of the hydrothermal extract. As a consequence, the same sample was also submitted as it is to UHPLC-IM-Q-TOF-MS analysis.
3.3. UHPLC-IM-Q-TOF-MS analysis
The freeze-dried extract obtained after HT was also analysed by UHPLC-IM-Q-TOF-MS, dissolving it in water (see Fig. 1). This approach was applied to characterize in detail various degradation products with higher Mw that may originate from the thermal process. To the best of our knowledge, this is the first time that such a technique is used for this purpose. The sample was analysed in triplicate both in positive and in negative ion modes to enlarge the metabolite coverage. At first, 4852 and 3941 features were aligned in ESI+ and ESI− modes, respectively, and among these features a total of 212 compounds were putatively annotated. The selection of the compounds of interest was carried out at first checking their abundance and their fragmentation score. Then, only those compounds whose experimental fragmentation pattern matched with the theoretical and/or with the one present in the online databases were retained. Finally, duplicate metabolites such as those ionizing in both polarities were checked. Some features with even good abundance but low fragmentation score have therefore not been considered, even though they could constitute compounds with relevance for bioactivity or risk assessment. The workflow applied is represented in Fig. 2. As a further confirmation, m/z values of each compound were plotted against experimental TWIMS CCS (Fig. S4 of the supplementary material). The compounds presenting a mismatch between m/z and CCS have been reported as marked with an asterisk within the table containing the whole list of annotations (Table S1, supplementary material). These ions with low m/z values and high CCS may be due to post-mobility fragmentation. They may correspond to fragments formed after the TWIMS separation due to the low collision energy of 6 V applied to ensure a proper ion transmission through the collision cell. Most of these ions are annotated as acids and they could be originated from higher m/z value compounds such as acyl-quinic acids.
 Food Chemistry Volume 421, 2023, 136150: Fig. 2. Representation of funnel filtration of the compounds identified by UHPLC-IM-QTOF-MS.
Food Chemistry Volume 421, 2023, 136150: Fig. 2. Representation of funnel filtration of the compounds identified by UHPLC-IM-QTOF-MS.
Several examples of identification confirmation are reported in Fig. S1 of the supplementary material. The whole list of putatively identified compounds and their respective chemical structures are available in Table S1 of the supplementary material. All the selected compounds were finally classified in chemical classes, according to the main functional group. The inclusion of a compound in a specific class was in some cases arbitrary, being many molecules complex structures with more than one functional group. However, the secondary functional groups have been specified in Table S1.
Overall, all the compounds were classified into twenty-two classes, as reported in Fig. 3.
 Food Chemistry Volume 421, 2023, 136150: Fig. 3. Distribution of the compounds identified in different chemical classes, in terms of number of compounds per class (a) and relative abundance (b). Relative abundances were calculated dividing the peak area of each metabolite by the sum of the areas of all the metabolites annotated in that sample.
Food Chemistry Volume 421, 2023, 136150: Fig. 3. Distribution of the compounds identified in different chemical classes, in terms of number of compounds per class (a) and relative abundance (b). Relative abundances were calculated dividing the peak area of each metabolite by the sum of the areas of all the metabolites annotated in that sample.
Some classes of compounds ionized both in ESI- and ESI+, and they are anhydro oligosaccharides, ketodeoxyoctonic acids (kdos), deoxy oligosaccharides, sugars conjugated with a large range of phenols, sugar conjugated with phenyl/diphenyl groups, sugars conjugated with various compounds (as for example pyran, fatty acid, polyol, ketone, purine), then organic acids, aldehydes, phenols, benzene-containing compounds, peptides, ketones, and furans. Then, in ESI- analysis one fatty acid and some pyran-derivatives were also detected, whilst carrying out ESI+ analysis even some sugar pyrimidinones, amino sugars, polyols and esters were comprised in the list. It is important to specify that many oligosaccharides and acetylated sugars were also putatively identified, but not selected nor reported in the list because not definable as degradation compounds. Indeed, both acetylated or unsubstituted sugars and oligosaccharides are formed from the hydrothermal extraction of hemicellulose, and they usually are the target compounds of this kind of process.
Generally, it can be noted that the main classes of molecules previously identified by GC–MS (i.e., organic acids, phenols, polyols, fatty acids, and modified sugars) came out with UHPLC-IM-Q-TOF-MS as well. As expected, many different types of modified sugars were detected, each of them with degrees of polymerization ranging from 2 to a maximum of 8 (Table S1). Each of the seven anhydro oligosaccharides was identified to be built up by agarobiose residues, and therefore they can be classified as agaro-oligosaccharides (AOS). Here, different DPs were detected, ranging from 2 (neoagarobiose) to 6. Agar/agarose is usually extracted from red seaweeds and basing on our knowledge no reports of its presence within lignocellulosic biomasses are present in literature. For this reason, their origin may need to be found from galactose degradation following the high temperatures employed in the HT. A recent work has proposed the mechanism of formation of levoglucosan and cellobiosan (i.e., glucose-based anhydro sugars) starting from cellulose by pyrolysis through hydrolysis and transglycosylation reactions (Leng et al., 2018), thus this may suggest the presence of galactose in HS in the form of galactan polymers. Seven compounds were also found and classified as Kdo sugars, which means that contain at least one 3-deoxy-d-manno-oct-2-ulosonic acid (or ketodeoxyoctonic acid, Kdo) molecule. Although Kdo is often just considered a characteristic component of bacterial lipopolysaccharide, it was also found to be a component of the cell walls of higher plants and in particular it is present only in the rhamnogalacturonan II structure (Temple, Saez-Aguayo, Reyes, & Orellana, 2016). This may suggest the formation of these compounds starting from pectin fractions, and this may be confirmed by the presence in relevant amounts of galacturonic acid among the total monosaccharides of HS (Table 1). Indeed, the UHPLC-IM-Q-TOF-MS analysis revealed the presence of Kdo sugars with different structures and degrees of polymerization, in some cases bound to each other, in others linked to monosaccharides or sometimes even to deoxy-sugars or dehydro-sugars. Some structures are reported in Fig. S2 of the supplementary material. In general, all these compounds (Kdos, anhydro oligosaccharides, deoxy oligosaccharides) accounted for about 8% of the total compounds identified (Fig. 3). Among them, deoxy sugars were the most abundant in terms of ion intensity, albeit numerically less. Although this value cannot be considered totally accurate from a quantitative point of view, it gives an indication that it is likely that other classes of molecules are more present in the hydrolysate. In fact, apart from the aforementioned compounds, coming from hydrolysis and modification of cellulose and hemicellulose, many other molecules were detected. Among them, organic acids, which represented almost 5% of the total number and only 1.3% in terms of relative abundance. Within this group, it can be noted that none of the molecules detected by GC–MS or 1H NMR were found here, and this could be ascribable to the difference in their molecular weights. Indeed, all the organic acids identified by GC–MS or 1H NMR had a low Mw, with a maximum of 132 g/mol, while the ones detected by UHPLC-IM-Q-TOF-MS had Mw in the range 188 – 290 g/mol. Within this class, monocarboxylic (e.g., 2-deoxypentonic acid, quinic acid), dicarboxylic (e.g. 3-oxoheptanedioic acid) and tri-carboxylic acids (1-Butene-1,2,4-tricarboxylic acid and 1-Hexene-1,2,6-tricarboxylic acid), but also more complex structures have been putatively identified. For example, the carboxylic group was also found in complex molecules, containing other functional groups and cyclic structures in the carbon skeleton, as a propanoic acid containing an indene-dioxol group, a methyl-butenoic acid substituted with and epoxycyclohexenoic acid, a 4-oxo-pentanoic acid containing a cyclopentane ring. The complexity of these structures makes any hypothesis about their formation speculative, also considering the lack of analytical standards. However, their presence in the hydrothermal extract suggests the need to reach a better level of understanding about the complex molecular profile of thermally treated biomasses.
Another class of compounds which confirms the result obtained with GC–MS and 1H NMR involves lignin-derived aromatic molecules. Several sub-classes can be identified in it, depending on the functional groups and the molecular residues present bound together. Specifically, these sub-classes were phenols, sugar phenols (i.e., phenols conjugated with mono-/oligosaccharides), phenyls, phenyls/diphenyls sugars (i.e., phenyls or diphenyls conjugated with mono-/oligosaccharides). The most present in this sense were by far phenols and sugar-phenols, comprising in total 93 compounds out of 212, corresponding to 44% of the total number of compounds and to 63% of the total relative ion abundance. These compounds come from complex mechanisms involving lignin. Compared to the aromatic molecules identified through GC–MS analysis, the ones found here were once again of greater complexity and with a Mw on average greater, also because constituted by complexes between phenols and other molecules. In fact, as reported in Table S1, very different molecules were found within the phenolic class, for instance containing functional groups typical of anthraquinones, naphtalenones, anthracenes, benzoquinones, stilbenes, tetraphenes, and again sulphate groups, furans and so on, depending on the number of rings that were present in the structure and on how they were bound together. Most of the features identified have never been previously reported for hydrothermal extracts of lignocellulosic materials, therefore it is difficult to extract information about their mechanism of formation, potential toxicity or, on the other hand, beneficial effects. Only few molecules among the identified ones were previously discussed in the literature. The presence of these compounds mainly in natural sources suggests that they (or some analogues) probably derive from the raw material. For example, a good relative abundance was detected for C-2′-decoumaroyl-aloeresin G, a phenolic glycoside previously isolated from Aloe vera (Dias & Rauter, 2015). Then, 6,8-Dihydroxy-3,4-bis(hydroxymethyl)-1H-isochromen-1-one is an isocoumarin previously obtained from a sponge-derived fungus on the genus Penicillium (Qi et al., 2013), and in the same way (2R,3S,4aS,9aR,10R)-2,3,5,8-Tetrahydroxy-6,10-dimethoxy-3-methyl-1,3,4,4a,9a,10-hexahydro-9(2H)-anthracenone and methyl (1S,2S,3S)-2,3,8-trihydroxy-3-methyl-9-oxo-2,3,4,9-tetrahydro-1H-xanthene-1-carboxylate were associated with fungi in literature as well (Yang et al., 2012, Zhang et al., 2012). Chlorogenic acid butyl ester was previously isolated from the leaves of a species of tree and flowering plant in the family Anacardiaceae (Corthout, Pieters, Claeys, Berghe, & Vlietinck, 1992). 5,7-Dihydroxy-2-(4-methoxyphenyl)-8-(3-methyl-2-buten-1-yl)-4-oxo-4H-chromen-3-yl-6-deoxy-β-d-mannopyranoside, better known as icariside II, is a prenylated flavonoidglycoside generally found in Epimedii herbs (Szabó, Pál, & Dulf, 2022). On the other hand, Fig. 4 reports as an example the chemical structures of two putatively identified phenolic compounds not found in natural sources. They correspond to complex cyclic molecules, having Mw ranging from 328 to 468 g/mol and despite no specific information about their mechanism of formation is reported, the presence of coumaryl and sinapyl alcohols’ skeletons suggest their direct formation from lignin cleavage and intramolecular recondensation. At the same time, some of these structures seem distantly reminiscent of the skeleton of some diarylheptanoids. These molecules are frequently found in plants belonging to the Betulaceae family and have been described in depth in a recent review about hazelnut’s phytochemicals (Bottone et al., 2019).
FIG 4
On the other hand, compounds carrying simple phenyl groups were numerically much less, and this is in line with the lignin origin of the most of these neoformed compounds. Nineteen compounds were detected and classified as phenyls or phenyl sugars, depending on the eventual presence of sugars residues, and even some biphenyl sugars were putatively identified in the extract: this is not surprising, since it is a typical bond that can be found in lignin structure. As in the other classes of compounds, it can be noted how different combinations of chemical structures were present again, such as furan-containing or acetic acid-containing molecules, and how different sugar moieties can occur. In general, the aromatic compounds detected showed extraordinary structural complexity. In a recent study, Kim and colleagues evaluated which are the main reactions lignin undergoes during a heat treatment. The authors concluded that these reactions concerned a methoxyl groups cleavage in C3 and C5, a propane side chain cleavage, a cleavage of the β-O-4 bond (depolymerization) and some simultaneous condensation reactions between released fragments, causing high molecular weight lignin (Kim et al., 2014). Although in our study the temperature of the HT was a little bit lower, our findings suggest that most of these condensation reactions may have taken place. Moreover, in the aforementioned work the lignin was extracted and thermally treated as it was, while in our case many other compounds, mainly derived from hemicellulose, had the chance to interact with lignin fragments, making even more complex the reaction pathways.
Another predominant class both in terms of relative abundance and number of compounds corresponded to furans, and sixteen molecules were detected and classified in this category. The most abundant one in negative ion mode was found to be a furan complexed with a hydrocarbon and containing a thio group, namely 5-(Tetradecylsulfanyl)-2-furoic acid. Some furancarboxylic acids have been reported to derive from oxidation of HMF (An, Sun, & Xia, 2019) and the presence of sulfate may therefore be due to some previous Maillard reactions with sulfur-containing amino acids. A derivative of maleic anhydride, namely 3-(4-Methyl-2,5-dioxo-2,5-dihydro-3-furanyl)propanoic acid was also found. In general, the putatively identified furans were once again found within very complex molecular structures, namely linked to other fragments of molecules containing other functional groups, such as phenols, phenyls, organic acids, and fatty acids (Fig. S3 of the supplementary material). Interestingly, simple furans as HMF or furanoic acids have not been detected with any of the analytical techniques used in this work, likely due to a loss in the most volatile molecules during HT extract freeze-drying process. The basic mechanism of formation of these furan-based molecules starts from sugars dehydratation, both starting from hexoses to form 5-HMF and/or from pentoses to form furfural, also depending on temperature and pH. Then, they may react in different ways, as dienes in the Diels–Alder reaction, but also through radical, electrophilic or polymerization reactions. A recent interesting review has been published about furans mechanisms of reaction (Gandini et al., 2022).
4. Conclusions
Since hydrothermal treatment is a very commonly used method to extract hemicellulose from lignocellulosic biomasses, it is essential to deeply investigate which are the degradation compounds that are simultaneously formed. Most of the literature available so far employed GC–MS analysis to identify the molecules originated from this kind of extraction. However, this technique presents some limitations in detecting high molecular weight and more complex compounds that may result from the degradation, hydrolysis and condensation reactions between lignin, hemicellulose, and other constituents of the matrix. In this work, three different techniques, namely 1H NMR, GC–MS and UHPLC-IM-Q-TOF-MS have been employed to investigate and identify the degradation compounds after hydrothermal treatment of hazelnut shells, producing diverse but complementary information. These approaches led to the detection of some common chemical classes of compounds, likely the most abundant, such as organic acids, degraded/modified sugars and aromatic compounds, and 1H NMR permitted to quantify them too. The two chromatographic-MS based techniques also allowed to detect different classes of furans, polyols, N-heterocyclic compounds, aldehydes, ketones, esters, among others. However, deciphering the chemical mechanism involved in the formation of all these compounds is still a major challenge. On the one hand several variables, primarily the HT severity, in terms of time and temperature, strongly influence their formation. On the other hand, also the initial matrix plays a fundamental role, since the proportion and the structure of the various polymers within the lignocellulosic complex can have a huge impact on the reaction mechanisms and products. However, sophisticated techniques such as UHPLC-IM-Q-TOF-MS are emerging and enabling to bridge this gap, allowing researchers to create datasets like ours, which can hopefully be used for the construction of specific metabolomics/”reactomics” databases, so that more light can be shed for the understanding of the formation of all these compounds and also for the building of predictive models.
- A multiplatform metabolomics/reactomics approach as a powerful strategy to identify reaction compounds generated during hemicellulose hydrothermal extraction from agro-food biomasses. Andrea Fuso, Laura Righetti, Franco Rosso, Ginevra Rosso, Ileana Manera, Augusta Caligiani. Food Chemistry Volume 421, 2023, 136150. https://doi.org/10.1016/j.foodchem.2023.136150.